
研究テーマ
主要な研究テーマ
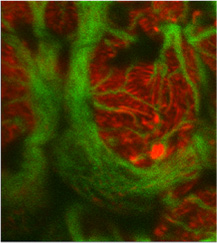
研究の主体は、原因に関わらない糸球体障害進展機序を明らかにすることです。つまり、糸球体という腎機能に最も重要な微小器官の恒常性の破綻と、それに対する糸球体内部環境の応答を明らかにすることで、腎障害因子が何であっても、その後に起きる糸球体の様々な変化(病変)を、形と分子でドキュメントすることで、どのようにして(how)糸球体が壊れるのか、そのルールを見つけることです。私たちがずっと注視しているのが、ポドサイトという細胞で、糸球体機能の中枢を担っています(ポドサイトについて)。
ポドサイトの研究は、現在世界的流行ともいえる勢いがありますが、私たちの研究室では、20年前からポドサイトの重要性を指摘し、病変の中から方向性を見出しながら研究を続けてきました。真実は病変の中にあるという病理のドグマ信じて、病変を観察しながらその中に疑問や課題を見出し、方法論を考え結果を論文として公表してきました。
科学研究は、論文の引用回数を目安に評価されますが、私たちの論文のいくつかは、世界に先駆けたものとして広く引用され、病理ならではの、" 現場(病変)の観察 " に基づいたユニークな研究として高く評価されています。私たちの目指すことは、腎障害機構に新しい視点(概念)を見出すことです。
そのいくつかを紹介しましょう。
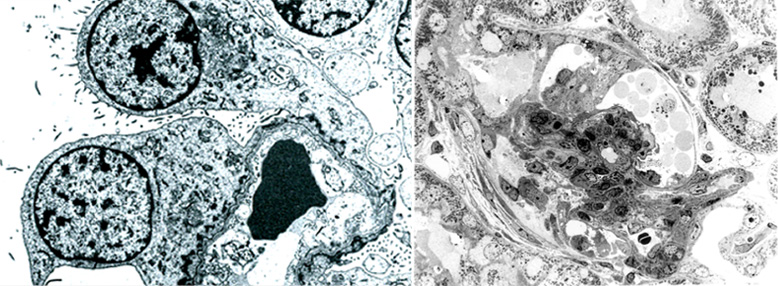
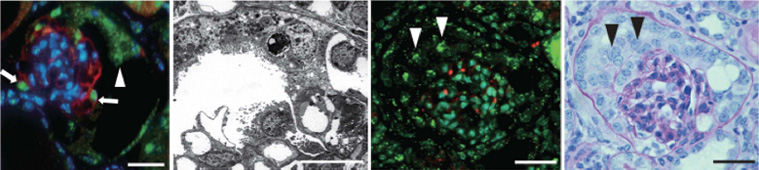
左図:フィンランド型先天性ネフローゼ症候群、電顕像。ポドサイト足突起が存在せずtight junctionがみられる。
右図:Collapsing FSGSマウス糸球体。壁細胞が増殖・遊走して基底膜を直接被覆している。
ポドサイトについて
腎臓の最大の機能は、血液の濾過と体外への水の排出です。人間には左右2個の腎臓があり、個人差はありますが、ひとつの腎臓当たり100万個の糸球体があります。糸球体は、血液から尿を濾しだす篩(ふるい)として機能しています。
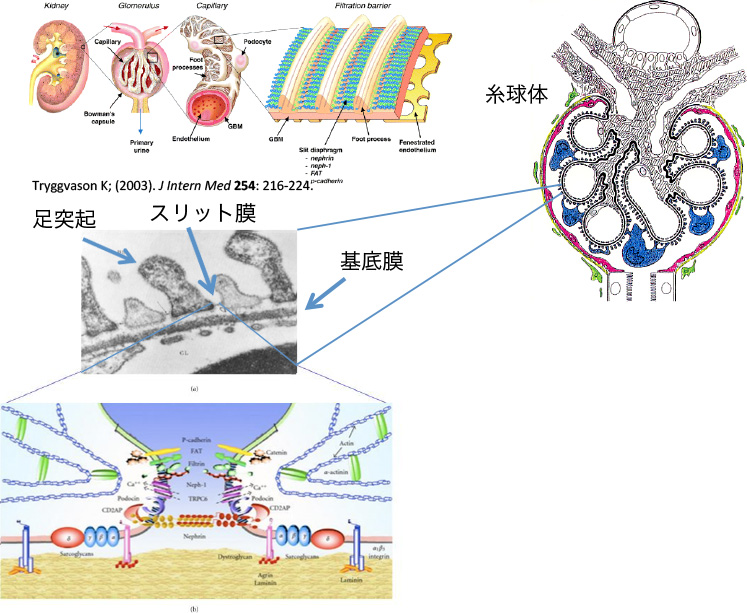
ポドサイトは、糸球体基底膜の外側を覆う大きな細胞(青)で、隣り合ったポドサイトと歯車のように趾が絡んでいます。これをinterdigitationと言いますが、ここに血液から蛋白を含まない原尿を正確に濾過する精巧な仕組みがあります。この趾の絡み合いの間にスリット膜といわれる多くの分子で構成される細胞接着装置があり、蛋白尿が漏れないようになっています。ポドサイトには、多くの機能があることが最近分かってきました。そして、そのいくつかの重要な分子に異常がおきると、ポドサイトの機能が低下し、蛋白尿の制御が出来なくなって、最終的には糸球体は壊れてしまうと考えられています。
ポドサイトに関する最近の研究は、その機能の多様性と背景にある分子のネットワークを明らかにしながら、ポドサイトの機能不全による糸球体障害を理解する、という方向性が主流です。私たちは、ポドサイトの機能的障害による蛋白尿発症に加えて、その終末像であるポドサイトの喪失と糸球体硬化の"how"に焦点を当てて研究しています。
ひと言でいえば、糸球体がポドサイトを失った場合、それを代償あるいは補填する反応が起こる(おそらく生体に不利な蛋白尿を止めようとする)という生物学的な現象の意味を理解することです。この現象こそが、糸球体硬化であり、その分子背景は、おそらくあらゆる糸球体障害に対する共通の生体防御機構であろうと考えています。ここには、複数の細胞や細胞外基質の間に、我々がまだ知らない秘密の会話(クロストーク)があるはずです。それを聴きだして糸球体病を理解する延長線上に、疾患の原因に関わらない治療の標的が見えてくると考えています。
1.ポドサイトが重要だと気付いたいきさつ
私がドイツのハイデルベルク大学に留学していた1990年のことです。その頃は誰もポドサイトには興味を示していませんでしたし、私もポドサイト研究をしようと思って行ったわけでもなく、またその研究室(Kriz教授のラボ)は、電子顕微鏡で尿細管やメサンギウムの正常構造を詳しく調べて、"機能形態学"、つまり形として見える合理性を機能の背景として論ずるという手法で研究していました。機能を司る分子の同定や、それを操作するという今のような方法論が殆どなかった時代です。
私は、当時流行っていた糸球体過剰濾過説に興味を持っていましたが、腎臓領域では最もインパクトの高い学説にもかかわらず、それによって糸球体がどのように壊れるか(how)の説明は一切ありませんでした。糸球体が過剰濾過により大きくなったら壊れるという2つの現象を結ぶルールを知りたかったのですが、当時1980年代後半はメサンギウム増殖全盛時代でしたから、糸球体過剰濾過による糸球体硬化はメサンギウム増殖で説明されようとしていました。しかし、こどもの腎生検を観る限り、メサンギウムなんて増殖しなくても糸球体は壊れると思っていました。
Kriz教授は走査電子顕微鏡が大好きで、私に大きくなった糸球体を観察したら、と促してくれました。本当は組織上の遺伝子発現を調べたいと思っていて、こんなに古くさい方法で、表面しか見なくて何が分かるのかなあ、とも思いました。でも、毎日暗室にこもって次々と映し出される立体的な像を観るのが楽しかったことはよく覚えています。大学への往復はLeimenという10km離れた街から、ドイツらしい田舎道を自転車で通いましたが、毎日新しく撮影したイメージを頭の中で弄びながらの通勤でした。
病理学というのは、起きた病変を遡って、最初にどこにスイッチが入るのかを調べる学問でもあります。動物に腎臓病を起こしても、腎臓の中にある糸球体の壊れ方は一斉でも均一でもなく、糸球体間に時相の差が生じます。
これを利用して、どのように壊れるのかという手順を知ることができるわけです。何にもなさそうで何となく変だなという糸球体の観察に傾注し、壊れる前の糸球体を注意深く観察していた時に、それまで“何も変わってないなあ”と思っていた大きくなった糸球体表面に、立体的であるはずのポドサイトが、平坦になって糸球体にべったり張り付いていることに気づきました(下図)。何度となく観てきたイメージの中に、突然新しいことに気が付くというのは形態学ではよく経験することです。
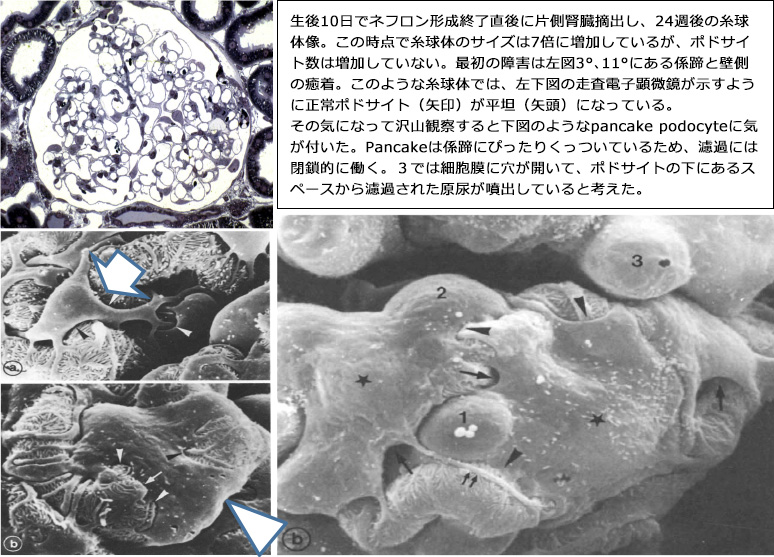

さて、このフラットになったポドサイトにKriz教授は少し興味を持って"Pancake podocyte"と名前を付け、"Machen Sie weiter, wirklich.."と言いました。もっとやってごらん、ということです。はじめて私がやっていることに興味を持ってくれたと感じ、頑張りたいと思いました。どうしてpancakeになるのか、その時はよくわかりませんでしたが、ある朝自転車で研究室に向かう途中クルミ(糸球体に似ている)を拾っていた時に、"糸球体が大きくなっても数が増えない、そしてポドサイトの足は基底膜にくぎ付けだから延びるかもしれない"と思って、糸球体が大きくなる前と後のポドサイトの数を調べたところ、糸球体は7倍に大きくなっていましたが、ポドサイトの数は変わっていないことが分かりました。このポドサイトの数を調べる方法も、当時はWT1などなく、Weibel. Stereological Methodsという、かなり難しい形態解析の本を渡され、普通はやらない家に帰ってからの勉強をして、算出のパラメータを決定し、300枚の糸球体の写真を大きく引き伸ばして数値を求めました。この時に、研究室にマッキントッシュのClassicというPCが一台あり、未来にやってきたという錯覚を覚えました。この使い方を、ラボのVon HargemannやRolfがとても親切に教えてくれました。
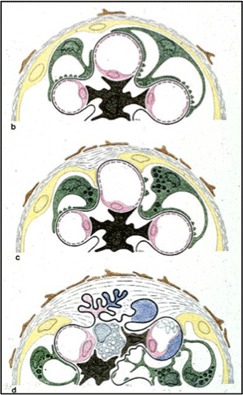
左図は、上記のモデルで観察したたくさんのイメージを、ひとつの絵にしたもので、Kriz ラボのRolfの作品です。
ポドサイトが脱落すると、むき出しの基底膜から蛋白尿が出ますが、それに対して、ボウマン嚢が反応し上皮あるいは基質が直接ポドサイトがなくなった基底膜を覆うという反応が起きます。しかし、おそらく蛋白尿漏出は止められない(ポドサイトではないから)ため、ボウマン嚢やその周囲に濾過された原尿がしみだして、線維芽細胞や基質の増加をきたして分節性に硬化するという仮説を提出しました(Nagata, Kriz, Kidney Int 1992, 引用201回)。
この現象は、特殊なモデルでのみ起きることではなく、どんな糸球体疾患でも起こりうる重要な機序だと思いましたが、Krizはそれを受け入れず、慎重に論文を直して投稿してくれました。この論文ともう一つ同時に、毛細血管の変化についてKidney Intに出したのですが(引用134回)、レフリーは2つの論文を一緒にして短い1論文にしろと言いましたが、Krizの説得で結局2つに分けて公表されました。(この2つの論文が短いひとつの論文になっていたら、現在の展開はなかったかもしれません。)
もう一つの幸運は、現在世界中でポドサイトマーカーとして使われているsynaptopodinが、私がポドサイトに眼を向けた時、丁度にKrizのラボの学生だったPeter Mundel(現ハーバード大教授)によってクローニングされたことです。これで、ラボの興味が一気にポドサイトに向きました。
今ではどの論文でも”podocyte is a highly differentiated post-mitotic cells and less capability to regenerate..”と書かれますが、調べてみると私たちが気づく10年くらい前に、Archen大学のPabstという人が、正常なラットの糸球体にthymidinを取り込む実験をして、ポドサイトにthymidinが取り込まれたのは2000個の糸球体の中たった1個だけだったと記載していました。当時はPub medなんてない時代でしたし、何かの学会の二次抄録だったので、ほとんど日の目を見ていなかったようでした。私たちよりもエレガントな方法で10年も前に明らかにしていたことに敬服しました。これが病気の原因だということに気付いたのは私たちが最初だったということに落ち着きました。その後2000年頃に盛んにポドサイトは増殖するという論文が出てきましたが、自分の眼で見たことから間違いだと思っていましたので、その仮説に加担しなかったことも結果としてはよかったと思います。
このようにいくつかの幸運や手助けがあって、ポドサイトの重要性に気が付いたわけです。もうひとつ、いつも生活の中でこのことを考えていたから、いろんなことが見えて来たように思います。イメージは見たい人にしか見えない、言い換えれば、そのイメージを待っている人に見えてくるものです。私にとってイメージを持つというのは、いつも愉しみであり、また新しいアイデアが浮かんでくるのも、意識の下にいつもイメージを遊ばせておく、というコンディショニングがあるためだと思っています。これは研究以外でも、気持ちを豊かにしてくれています。
2.私たちのポドサイト研究(1992-2004)
ポドサイトに偶然関心を持ち、それがイメージ通りに展開したことなどから、病理の世界に入ってきたようなものですし、現在の職もポドサイトが道を付けてくれました。帰国して臨床医として働きながら研究を続け、それが高じ病理医に転じた時にも、多くの先達や同僚から研究を続ける動機と機会を頂きました。これが現在、楽しく研究を続けられている最も大きな要因です。
新生児病理をやっていてよかった
さて、"ポドサイトは増えない"から次にイメージしたのは、ではいつ増えなくなってしまうのか?ということ。小児科医時代、新生児科で担当した極小未熟児が生後急性腎不全になり亡くなりました。当時病理学教室にも所属していたので、どうして尿が出ないのだろうという疑問を解きたかったわけですが、病理解剖で得られた腎臓のイメージを観て息をのみました。
これは、在胎21週で生まれてきた新生児ですが、表面に近い部分の糸球体が出来ていません。この未熟な糸球体の曲線が印象的で、まさにmetamorphosis(変態)というイメージです。
当時、PCNA(proliferating nuclear antigen)という細胞周期が活性化した時に核に発現する蛋白(DNA polymerase delta)に対する抗体で発現を観たところ、糸球体として成熟していないものには多数陽性細胞があり、毛細血管が備わったものには全く陽性細胞がありませんでした。ポドサイトは、濾過障壁としての機能を獲得したと推察される状態になった(capillary loop stage)に、分裂をストップして以後増殖はしないということ、さらにこの増殖を停止したことと、間葉細胞のマーカーであるビメンチンを発現したことが同時であったことから、ポドサイトの成熟(分化)には細胞周期の停止が必要だと結論しました。発生過程の腎臓は、形がダイナミックで背景の細胞の形質変換なども起きていて観ていて面白かったです。この論文が1993年に発表されてから、ポドサイトの研究には発生過程の観察が入れられる様になりました。思わぬところにアイデアのヒントがあるものです。ただ、こういう現象は細胞生物学や発生学ではよく知られていることだということも後ほどわかり、いろんな分野の勉強をする必要があるなとも思った次第です。ちなみに帰国して初めて自力で書いたこの論文は、地味な雑誌(Nagata et al. Anat Embryol, 1993、引用87回)に掲載されました。

再び標本がくれたアイデア
では、メサンギウム細胞や内皮細胞の増殖はよく知られているのに、ポドサイトの増殖は知られていないのは何故だろうと疑問を持ちました。そう思いながら生検標本を観ていたら、核が複数あるポドサイトが眼に入ってきました。これに気が付くと、多くの病気で多核のポドサイトがあることがわかりました。やはり疑問とイメージを持ちながら観察する、そして見えるものは説得性があります。そこで、核が多数になるためには1個の核が分裂するが、細胞が2つに分かれる(cytokinesis)ことができないのだろうと考えました。ならば核分裂をしているポドサイトがあるだろうと探したところ、170例の生検組織中たった1個、FSGSの症例でこれを発見しました。さらに、電子顕微鏡で2核のポドサイトを観たところ、足突起が消失していたことから、細胞が核分裂するときに一旦細胞骨格が消失し細胞が2つになる時に再構成されるのが、細胞が二個になれないために再構成できないと考え、ポドサイトの核分裂はmitotic catastropheとして細胞障害をおこすと結論し、簡単な論文にしました。
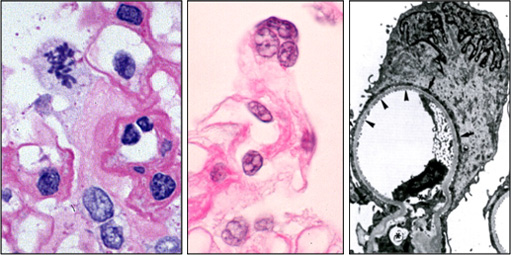
左:FSGS症例に観られたポドサイトの有糸分裂像。細胞質が広がり、基底膜を覆っている(足突起消失) (AnatoEmbryol 1995, 引用87)
中:腎炎患者に診られ多核ポドサイト。(Nagata et al. Nephron 1995, 引用48)
右:FGF-2毒性テストから発見された多核ポドサイトの足突起消失像
(Kriz et al. Kidney Int 1995、引用176)

Cyclin dependent kinase inhibitor(p27)
それ以来、ポドサイトの細胞周期を止める分子は何だろう?と思い続けていました。ある日、日本病理学会で、MayoクリニックのLloyd教授が、下垂体細胞が腫瘍化する時に、細胞周期の抑制が解除されるとして、p27という細胞周期抑制分子の話をしていました。細胞の腫瘍化をp27で判断、という話だったように思います。p27は、細胞周期を抑制する(細胞分裂を止める)分子として、1994年にToyoshimaらが同定しCell誌に報告した分子で、p27欠損マウスは臓器が肥大することもすぐに報告されました。Lloyd教授の話を聴きながら、発生期の腎臓でこのp27がポドサイトの細胞周期を止めているのではないかと思い、早速抗体を手に入れて増殖マーカーKi-67との供発現を組織上で観察しました。最初に顕微鏡を観た時にはまた、ドキッとしましたね。予想どおりだったからです。すぐに論文として投稿しました。嬉しかったのは、この論文の掲載誌の表紙に使われたことです(右)。
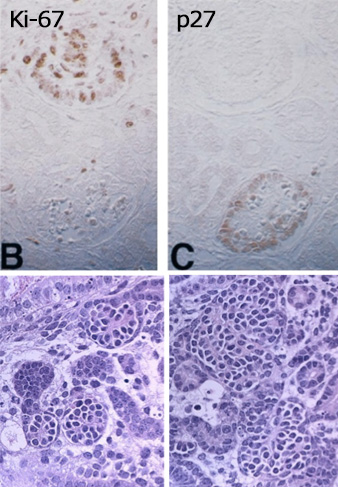
ヒト糸球体分化過程でのKi-67(B), p27(C)の発現を連続切片を観察すると、糸球体の分化が進むとこの2つの細胞周期関連分子の発現が鏡面的(mirror image)であることがわかる。
つまり、p27が発現することで増殖が停止するということを示している。(Nagata et al. Am J Pathol, 1998、引用110)
それでは、p27がポドサイトの増殖停止と分化を決定しているかと言えば、そう簡単ではないことがp27が欠損したマウスでも糸球体ができることからわかりました。おそらくp27は類似した分子(p57, p21)によって代償されるのではないかと考え、九州大学の中山敬一先生が開発されたp27+p57ダブルノックアウトマウスを調べさせていただいたところ、ダブルノックアウトマウスでは、糸球体が面積として4倍くらい大きいのです。これにもビックリしました(左下右図)。そしてポドサイトの数も約3倍もあることが分かりました。分化については、WT1やSynaptopodinといったポドサイトの分化マーカーの発現はあり、スリット膜も形成されていたのでこれらの分子が分化に直接大きな影響をしていることはないものの、糸球体のサイズやポドサイトの数を規定している可能性が考えられました。本当は大発見なのですが、学生の卒業もあり簡単に掲載される雑誌を選びました。
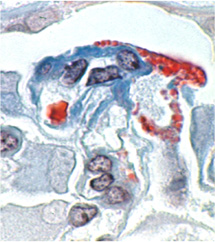
初めて糖尿病とポドサイトの関係を明らかにした論文
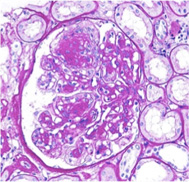
糖尿病性腎症は、現在世界中で増加の一途をたどり、慢性腎不全の最も頻度の高い原因です。糖尿病性腎症は、糖尿病の患者さんに多量の蛋白尿と腎機能低下がみられ、腎生検で右図のような特徴的な像が確認できて、はじめて診断できます。この病理組織像は、有名なKimmelstiel Wilson結節と言われており、本態は糸球体のメサンギウム基質の増加である考えられています(右図)。1990年代には、メサンギウム細胞を培養皿の中で増殖させたり、細胞外基質を産生させる実験が流行しました。これは方法も簡単で、細胞もよく反応するため実に多くの論文が出ました(今ではほとんど引用されることはありません)。メサンギウム細胞に糖や、糖の代謝産物を添加すると、1-2日で細胞が増殖するので、糖尿病性腎症では長期に糖に暴露されることでメサンギウム細胞が細胞外基質を作るという筋書き通りの結果が相次いで報告されました。
筑波大学に赴任して数年して、糖尿病性腎症の研究をしたいと大学院にやってきた星さんは、製薬会社の研究員で、アメリカから自然発症ラット糖尿病モデル(ZFDfa/faラット)を輸入して研究していたので、そのラットの腎症を病理学的に研究することにしました。それまで、動物の膵臓を薬剤で破壊して糖尿病を発症させる研究報告はありましたが、どれを観てもヒトの糖尿病性腎症とは似てないにもかかわらず、メサンギウム増殖病変ができ、ヒト腎症に似ているという結論が出ていて変だなと思っていました。当時JCIというアメリカの権威ある雑誌に、糖尿病性腎症PKC説というのが発表され、糖によってPKCという細胞内シグナルが活性化し、メサンギウム細胞の増殖が起きると提唱されていました。星さんとその会社の上司は、このラットもメサンギウム病変を見つけ出そうと考えていました。このラットは30週を超えると顕著な腎臓病を発症し、病変を観察すると下図のような変化がありました。
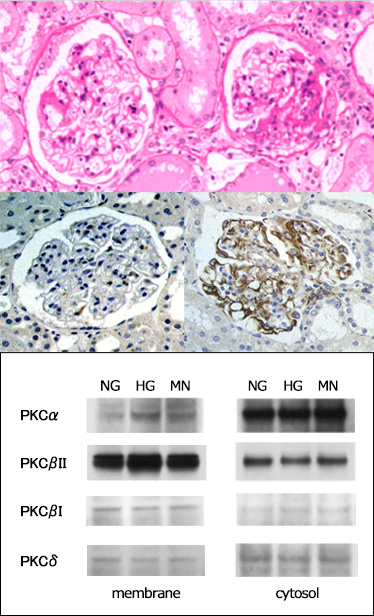
やはりどう見ても上図のヒトの糖尿病腎症の糸球体には似ていません。私は、これでメサンギウム増殖に焦点を当てて研究するのは難しいと考えました。同時に右側の糸球体の壊れ方が、ポドサイト障害による糸球体硬化とよく似ていることに気づき、糖尿病でポドサイト障害が起きる、ということを証明することにしました。すると、糖尿病モデルでは、糸球体が壊れる前にポドサイトにデスミンが発現していることが分かりました。つまりポドサイト障害が硬化病変に先立って起きるということです。
上記のPCK説に関しても、メサンギウム増殖に都合よく使われていましたが、同じことは他の細胞でも起きると考え、培養ポドサイトでのPKCを観ることにしました。私は生化学的手法に慣れていませんでしたが、星さんは実に上手で、糖負荷によりPKC-beta IIが活性化することと同時に、ポドサイトに細胞肥大と細胞周期抑制因子の発現がみられることを示しました。つまり、糖によりポドサイトにストレスが加わり、糸球体硬化すると結論しました。
ポドサイトと糖尿病というのは、当時は関連あるとは誰も考えていなかったし、学会で発表しても全然ウケなかったのですが、確信があったので、思い切って当時私の目標誌のひとつだった、米国の病理学会雑誌Laboratory Investigationに投稿し、困難なく掲載されました。
今では、ポドサイト障害は糖尿病性腎症の進展因子であると考えられていますが、これらの仕事は世界で最初にこれを示したものです。(Hoshi S, et al, Lab Invest, 2002, 引用209.)これは私のラボから出した論文の中では最も多く引用されています。同時に細胞実験のデータをBBRCに投稿し、一発受理(reviseなし)でした。(Hoshi et al. BBRC, 2002, 引用129)。ちょっと快心な仕事でした。図下は、糖負荷によりPKCbßIIが細胞質から膜へ移動することを示したもの。
日本で最初のCollapsing FSGS例
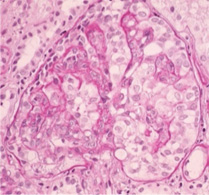
今でこそよく知られているcollapsing FSGSですが、大学内のカンファレンスで初めて見た時には、不思議に美しい病変だと思いました(右図)。
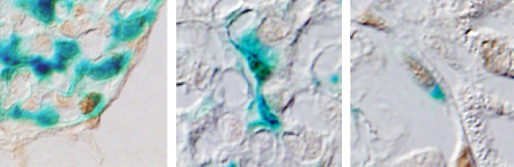
これは、栃木県の病院から診断ができないとカンファレンスに出された症例です。高度のネフローゼ症候群ですでに腎機能低下がある13歳女児でした。明らかに尿腔内に上皮系の細胞が増えています。これまでの経緯からポドサイトは増えないと信じていたため、この細胞は他の細胞だろうと思いマーカーで調べたところ、サイトケラチンが陽性で、ポドカリキシンが陰性でした。中に1個だけ両方陽性の細胞が観察され、ポドサイトと壁側上皮の間に細胞形質変換が起こっているのだと考えました(下図)。このイメージがその後のいくつかの仕事に発展します。
この頃"dysregulated podocyte phenotype with proliferation…"という内容の論文が、Kriz, D’Agati, MundelらからJASN(引用507)に出て、一大学説となりました。これに迎合するような論文がどんどん出てきた結果、ポドサイトは分裂増殖しうる、あるいは半月体の構成要素であるとしながら暗に増殖するとを述べ、ポドサイトパチーという概念が作られました。とくにHIVによって増殖する、これがcollapsing FSGSである、というものです。ここで、論理の微妙なスキップ、つまり"ポドサイトが増殖するときには細胞マーカーを喪失する、だからポドサイトのマーカーが発現しない"、がありましたが、これに対する批判は受け入れられませんでした。このポドサイトが増殖することに異を唱えるグループが2つ。私たちとオランダのDjikmannのグループでした。その後私たちは、この説の欠点である細胞起源の証明ができないことを、遺伝子操作技術により乗り越え、増殖しているのはポドサイトではないことを明らかにしました。この間、この2グループの論文は些か冷たくされましたが、病変をよく見ているという自負があり、またいろいろな方面から確信が持てたことで、その学説に迎合しようとは思いませんでした。この点でも、視たものを信じる病理学の基本に守られたと思っています。そのオランダのグループのSmeetsとは今もとてもいい関係です。
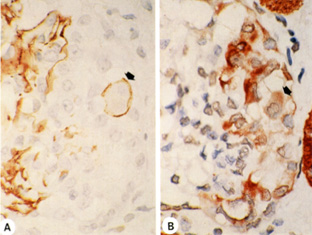
左図は、上記のcollapsing FSGS例の連続切片にてPHM5(ポドカリキシン、ポドサイトマーカー)とサイトケラチン(ボウマン嚢上皮マーカー)を染色したものです。多くのサイトケラチン陽性細胞の中に、ひとつだけ、両方のマーカーを発現する細胞がみられました。この細胞はなんだろうと思っていましたが、その後いろいろと調べてみると、かなり稀な現象だということがわかりました。とりあえず、ポドサイトは増えるとする説が定説になることにブレーキを掛ける意味で簡単な論文にしました。ポドサイト形質変換の概念の最初の報告です(Nagata et al, Am J Kid Dis, 1998, 引用59回)。
冷蔵庫に眠っていた古い血清がもたらしてくれた幸運
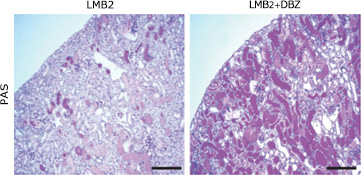
私たちは、初めてcollapsing FSGSではポドサイトは増えないことを遺伝子タグ付マウスによって証明しました。2009年のことです。この研究にもいくつかの幸運がありました。1999年にKidney Intに発表されたp21遺伝子欠損マウスに抗GBM抗体を投与するとcollapsing FSGSによく似た病変ができることを参考に、このマウスにポドサイトだけをネフリンプロモーターでbeta-GALによって青くできるマウスと交配し、恩師の渡辺照男先生が随分昔に佐賀医大で別の目的で作られて筑波に持ってこられ、研究室のフリーザーに眠り続けていた抗アヒル・ウサギGBM抗体を投与したところ、Collapsing FSGSが出来ました。ポドサイトを追跡したところ増殖細胞では一切ポドサイトの遺伝子タグは発現しなかったこと、さらにこれらのほぼすべてが壁側細胞マーカーであるクローディン1を発現したことから、collapsing FSGSではポドサイトが急に失われ、それに対する反応として壁側細胞が増える、という結論に至り、あるジャーナルに投稿しましたが"Although the result is clear, there is no relevance with human collapsing FSGS…"とされて即刻reject。すぐに違う雑誌に投稿したところ、"the data are very convincing…"として少し訂正してすぐに掲載されました。この2つの雑誌のレベルはあまり変わらないとは思いますが、この時には件のポドサイト増殖説が信じられていたから、査読者として腎臓関連の人が多い雑誌と、病理関連が多い雑誌との違いなのだろうと思いました。この教訓は、今投稿するジャーナルを考えるときに役に立っています。この仕事をしていた時も、渡辺先生が別の目的で作成された抗体が偶々あり、多くの人が破棄する機会があったのにフリーザーの中にちゃんと残しておいてくれた幸運を感じました。
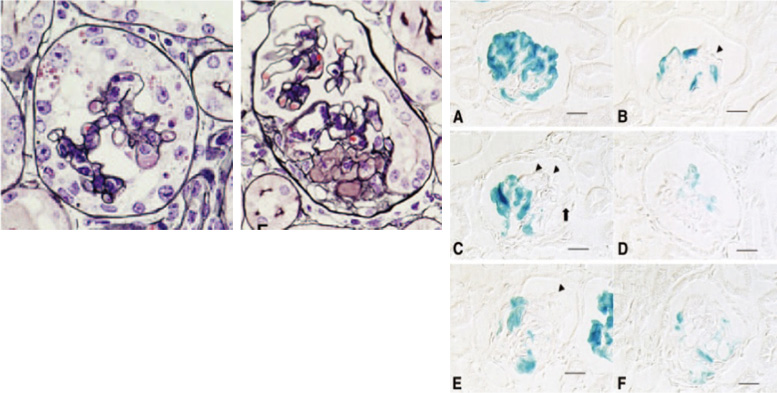
上図:抗ウサギアヒル腎粥投与p21欠損マウスにみられたcollapsing FSGS。毛細血管の虚脱と尿腔に多くの上皮細胞が増殖しています。マウスは重症ネフローゼ症候群を発症。
右図:nephrinプロモーターでポドサイトだけを青くタグ付した標本。Aは正常糸球体。ポドサイトだけが鮮やかな青色を呈しています。B, C, D, Eと病変が進むにつれて青色の細胞は減少していきます。B, Cではとくに上皮細胞は増殖しているのに(矢印)その細胞は青くはない、つまりポドサイト以外の細胞である、ということを明確に示しています。(Suzuki et al. Am J Pathol, 2009)
Diffuse mesangial sclerosisから学んだこと
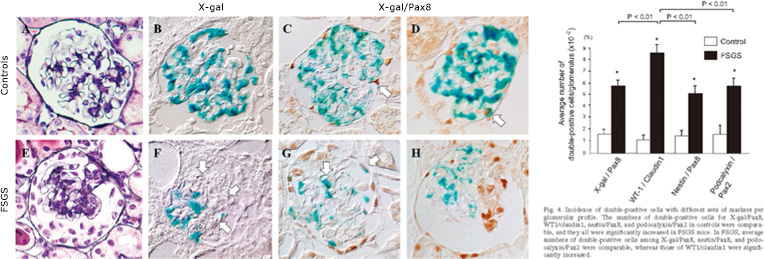
ポドサイトが増えるとか増えないとかという論争で、最も手ごわかったのがdiffuse mesangial sclerosisという病気の存在です。このDMSは、ポドサイトの分化に大変重要とされるWilms tumor suppressor protein-1(WT1)遺伝子異常により発病するとされています。WT1は、腎臓の形成にも重要で、同時に細胞増殖に関係するがん遺伝子でもあります。ポドサイトが増えると主張するグループは、このWT1の異常によってポドサイトが増殖するというシナリヲをつくり、podocytopathy説を補強していました。新潟大学小児科の池住洋平先生は、WT1遺伝子変異のあるDMS症例を2例経験し、まず病理診断について連絡をくれました。腎生検では、増殖している上皮細胞はポドサイトとも壁細胞とも区別はつきません(下図)。マーカー染色では、Claudin1 (+), Synaptopodin (-) であり、ポドサイトではないことがわかります。実はこれまでの研究では、壁細胞の可能性については、無視されていました(よく査読者が許したものだと)。尿中のポドサイト数が増加していることが確認され、ポドサイトが剥離する、その結果壁細胞が増殖すると結論しました。これは、私が待ち望んでいたドキュメントで大変嬉しかったです。やはり病理の雑誌に投稿し、あまり困難もなく掲載されました。このように、症例から考えたことを実際に症例で解析し発表することは、とても楽しい作業であり、病理と臨床医とのコラボの大切さを示す例です。
この論文で、we believe that there may be a similar mechanism with collapsing FSGS in which PECs proliferate predominantly in the lesions along with podocyte loss.と記しましたが、病理所見としてのメサンギウム増殖など明らかに異なる部分もあるため、さらに解析する必要があると考えています。
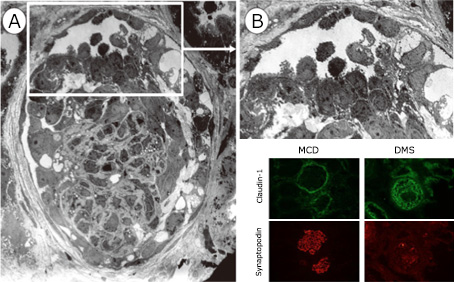
左図:DMS症例の電顕セミシン切片。
毛細血管が虚脱するだけではなく、メサンギウムが増殖しているように見えます。Bでは上皮細胞が増殖しており、その細胞はポドサイトのマーカーは陰性で壁細胞のマーカーが陽性です。そして、この論文のいいところは、尿中にポドサイトが大量に出ていることを示したことです。
(Ikezumi et al. Human Pathol 2014)
3.最近考えていること:糸球体微小環境の恒常性
スリット膜関連分子の同定や、家族性FSGS症例での遺伝子検索がポドサイト機能に重要であるとする多くの報告は、糸球体機能の恒常性を維持する機構を理解するのに重要な意味があります。糸球体機能は、血流の維持、透過性の調節が基本ですが、そのためには、糸球体毛細血管内の微小循環の恒常性が重要です。ポドサイト、糸球体基底膜、内皮細胞は濾過障壁というユニットを構成しています。濾過のバリアである基底膜は、ポドサイトと内皮細胞から常に産生され崩壊しながら、そのかたちと機能(量と質)を維持していると考えられています。これは、双方の細胞が何らかのシグナルを交換し、相互の作用を調節していると考えられます。特に、ポドサイトが産生するVEGFに対する受容体を内皮細胞が持ち、VEGFは内皮細胞の生存に重要であることから、このシグナルの候補としてはVEGFが最も可能性が高いとされています。内皮細胞とポドサイトがどんな会話をしているのか、聴いてみたくなりますが、まずこれを明らかにするためには、お互いがどのように依存しているのか、すなわち片方の細胞を選択的に障害することでもう一方の細胞の状態がどのように変わるのか、個体の中で可視化することが病理学の醍醐味です。生体には代償機構が沢山働くと考えられますが、一方の細胞の障害によって代償できない因子は、別の言い方をすれば、恒常性維持に決定的であるとも考えられます。私たちの最近の研究は、病変形成の機序の背景にある恒常性維持の仕組みにも広がってきています。分からないことだらけですが、カタチとして見えない分子を可視化して生体の機能を理解することは大変面白いです。
4.ポドサイトが障害されると次に何が起こる?新しい研究成果
東海大学の松阪泰二先生は、この数年来の最も親しい共同研究者です。彼が作製したNEP25マウスは、ポドサイトだけが障害されるモデルとして高いインパクトを以て2004年に発表されました。このマウスの最大限のメリットである“ポドサイト選択的障害とタグ付け”を利用した研究は、ヒトの標本では不可能な細胞の起源が追跡可能であり、これを武器に現在までいくつもの共同研究を行っています。
① ポドサイト喪失による壁細胞の応答の背景分子:Notch
結論は、FSGSはポドサイトを失った糸球体のNotch1による瘢痕修復作業の結果である、というものです。
これは、飯塚病院から大学院として入学した上野智敏さんが熱心に取り組んでくれ、最後のシグナル阻害実験では、またまた驚きのイメージを得ることができ、Kidney Intに掲載されました。
この論文では、ポドサイトが選択的に、しかも急激に(大量に)喪失されると、壁細胞にもともと発現していないNotch1という分子が発現し、これが細胞遊走のプロモーターになっているということを示しました。Notch1は、細胞増殖、分化、遊走など細胞がアクティブに動くときに一過性に発現する分子として知られています。まず、発現をポドサイト特異的障害モデルで調べたところ、壁細胞への発現がダイナミックに確認され、壁細胞に絞って研究を進めることにしました。その結果、Notch1のシグナルは、壁細胞に異常発現して壁細胞の遊走に強い働きがある、そしてNotch1を阻害すると、蛋白尿や腎機能が急激に悪化することが分かり、壁細胞はNotch1を発現することでポドサイトを失った濾過障壁を増殖と遊走と接着により修復し、それがFSGS共通の機序だろうと結論しました。しかし、ポドサイトが障害されると何故Notch1が壁細胞に発現するかは分かっていないので、さらに考えたいと思います。このように、病変で確認して確信をもって次進むことは実に病理的で楽しい作業です。
左上:NEP25マウスのポドサイト、壁側上皮にNotch1が発現し、電顕では、ひとつのPECが毛細血管に極性を変えて遊走している図が電顕でみられます(左図)。増殖したPECにはびまん性にNotch1が発現し(右図)、さらにNotch阻害薬により、蛋白尿は増加し、糸球体病変も進行していることが分かりました(下図)。つまり、Notch1はポドサイトを失った濾過障壁の修復を司る分子と考えられます。(Ueno et al. Kidney Int 2013)
② FSGSでのポドサイトと壁細胞間の形質変換:やはりポドサイト→壁細胞
これは、Notch1の上野さんの後輩で、同じく飯塚病院腎臓内科医であった坂本和雄さんが、腎臓学会の研究支援プログラムに採択され(実は私が委員長なんですが)、6か月間本当に頑張った成果です。
実験期間はわずか6か月でしたが、首尾よくいい論文にできました。
この仕事のアイデアは、上に紹介したcollapsing FSGSの症例で、増殖細胞の起源を壁細胞と結論して1998年に報告した時にヒットしたものです。沢山増えている細胞の中に、ポドサイトと壁細胞両方のマーカーが同時に陽性である細胞が1個見つかったことが、この2つの細胞間に形質変換あるいは移行があるのではないかという着想になったのですが、そうであれば2個の細胞が、どちらからどちらのへ変換するのか、その頻度がどのくらいなのか、そして実際に病変形成に関係しているのか、などまったくわからないまま議論されていたことにフラストレーションを持っていました。これまでの研究の大きなハードルは、細胞形質変換が起きてしまうと、どの細胞由来であるのか、細胞マーカーで判断することが出来なくなることです。とくにヒトではこれは決定的な限界となるので、実験動物を使って証明することにしました。ポドサイト由来である証拠としてのタグ付けをして、一旦ポドサイトになったら、その後どんな細胞に変わっても、青いタグでポドサイト由来だと判定できる、ROSA-Cre NEP25モデルのメリットを最大限利用しましました。
さて、問題はタグで由来を明確にして、同時にマーカーで分化状況を検出する。このために少し工夫しました。マーカーは2種類、ポドサイトと壁細胞のものを用いますが、片方が細胞の核に、他方は細胞膜に発現するものの組み合わせを考えました。同じ部分に発現するのでは、酵素抗体法を使ってでは色で識別ができないからです。二重染色の条件設定はかなり面倒な場合があり、また染色性の質が低いと何を観ているのかわからなくなります。
この手の実験には、予定調和がない、つまりこうなるであろう、こうでなくてはならんというものがなく、実際出てきたデーターから素直に結論を出せるものなので、染色の質の担保と細胞の同定、算出など地味で確実な仕事がとても大事です。結論として、ポドサイトが急激に大量に障害されると、ポドサイトと壁細胞の形質を同時に持つ細胞が増加すること、その分化の方向性はポドサイトから壁細胞に向けてであり、どうも逆ではないこと、さらにこの形質変換という細胞反応は、硬化病変を形成する部位には存在しないことから、形質変換はポドサイトが障害を受けた糸球体の応答のひとつで、あくまで病変形成は従来存在していた壁細胞によると結論しました。この結果は、上記のNotch1の機能とあわせて、FSGSの病変を理解することに大変役立ちました。なにより、自分が長いこと持っていた疑問が解決できてとても嬉しかったです。坂本さんは、これで米国腎臓学会(ASN)ではなかなか採択されない口演をし、かつbest trainee awardを受賞しました。で、この仕事はあまり問題なくAm J Physiol, Renal Physiolに掲載されました。
上図:ポドサイトタグ付NEP25マウスのポドサイト、claudin1の発現。ポドサイト由来でClaudinを発現する細胞は稀ですが、FSGSでは増加していることが分かりました。
下図:ポドサイトタグPECマーカー共陽性細胞(Sakamoto et al. Am J Physiol, Renal Physiol 2014)
③ ポドサイトドミノ倒しという新しい概念:ポドサイトと糸球体内皮細胞間クロストーク
最近、クロストークというキーワードをよく耳にします。これは、生体内で異なった細胞が協調して機能している(助け合っている)ことを表していますが、これを明らかにする方法がなかなか見つからないのも現状でした。
本当は、腎生検病理診断を学びたいと希望して連絡をくれた、腎臓内科医の小林凡子さんに、私の持っていた一つの疑問を一緒に解いてもらうよう、大学院に入学してもらいました。私はFSGSに興味を持ってますが、その病変はしばしば血栓性微小血管症(TMA)と似ていることがあり、この2つの病気の類似点から、ポドサイトと内皮細胞のクロストークを考えようというのがテーマです。
つまり、FSGSの背景にTMAがある、あるいはその逆。
FSGSについてはポドサイト障害ばかりが話題にされてきました。一部では根強い内皮細胞障害説がありますが、それを裏付ける論文はありません。実は、上記のNotch1の実験中に、ポドサイト障害部位に血栓ができるという現象に気が付いていました。つまり、ポドサイトを壊したら、その下にある内皮細胞が障害され(あるいは生きていけなくなり)血管内凝固が進む、ここにシグナル伝達がある。アイデアは、やはり病変からやってきます。クロストークは、その現象を促す分子の特定をもって初めて認証できることです。これまでポドサイトから内皮細胞に向けたシグナルとしてはVEGFが有力で、実際VEGF抗体で治療したがん患者ではTMAが見られたことや、ポドサイト特異的にVEGFを欠損させるとTMAに似た病変ができることは知られていました。しかし、内皮細胞からポドサイトに向けてのシグナルは全く分かっていませんでした。少し漠然と始めた実験ですが、小林さんはいろいろ広く論文を読み、PAI-1にその可能性があるかもしれないと考えました。抗体で染色すると、ポドサイトの障害を起こした直後に、本来発現しないPAI-1が糸球体内皮細胞に発現することがわかりました。これには、最初はどの様な意味があるのか見当もつきませんでした、というより血液が凝固するので、当たり前かなとくらいにしか私は思っていなかったのですが、実際には血栓形成の随分前なので、別の意味かもしれないと思っていました。このPAI-1についていろいろと調べて、ポドサイトに向かって何らかの作用をするかもしれないと考え、PAI-1阻害薬を開発されていた東北大学の宮田敏男教授との共同研究が始まりました。つまり、ポドサイト障害に伴って発現した内皮細胞のPAI-1が、何らかの作用をしているのなら、その阻害薬によって変化に表れると考えました。結果NEP25マウスのポドサイト傷害は、PAI-1阻害薬投与によって抑制できることが分かりました。つまり、ポドサイトが障害されたために(おそらくVEGFの産生低下による)内皮細胞に何らかの障害が起こった結果として、PAI-1が内皮細胞から産生され、これがポドサイトに再び何らかの作用を起こしてポドサイトを脱落させるという仮説を立てました。
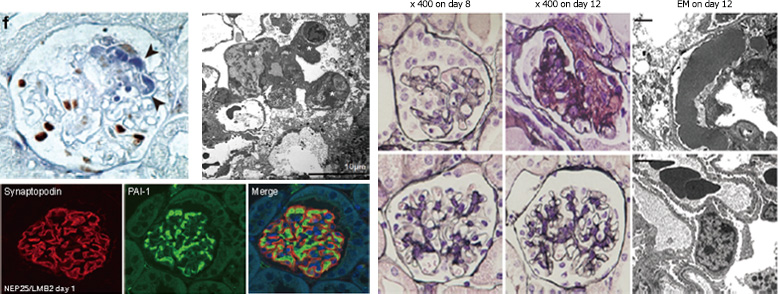
上左:NEP25マウスの血栓形成部位(矢頭)にはポドサイト(WT1+茶色)はみられない。上右:電子顕微鏡ではポドサイトのない部分に血栓がある。下:ポドサイトを障害すると血栓形成や蛋白尿が起きる前にPAI-1が内皮細胞で異所性発現する。
NEP25マウスにPAI-1阻害薬を投与すると、蛋白尿、血栓形成、さらにポドサイト数の減少が抑制された。
結果として、糸球体硬化病変も減少した。一方、電顕では、ポドサイトの足突起維持がみられる。(Koboyashi et al. Am J Physiol, Renal Physiol, 2015)
ひとつのボンヤリした仮説を検証することで、次の仮説が浮かび上がり、これを解くために研究を進めるという、研究の楽しさそのものの展開に大変心が躍りました。小林さんはアメリカ腎臓学会で2回oral presentationに採択されました。PAI-1がどうしてポドサイトを障害するのか、についても、癌の転移に関する論文を見つけて、おそらくインテグリンが失われるためにポドサイトの基底膜への接着維持ができないのではないかと考え、さらにuPA, u-PARと複合体を形成することで、インテグリンが細胞内にエンドサイトーシスされるために剥離するという仮説を、いろいろな方法を使って明確に示しました。最後のキーワードのエンドサイトーシスを証明するのは難しいので、internalizationという単語を使って濁していましたが、順天堂大学の栗原秀剛先生にご指導いただき、培養ポドサイトに二重免疫電顕をおこなってエンドサイトーシス小胞にインテグリンとu-PARが存在することをイメージとして提示することができ、これですべてが繋がりました。これは、ポドサイト障害の因子が何であれ、一旦障害されると内皮細胞からのPAI-1が、最初のポドサイトの障害因子とは関係なく(障害因子が除かれたとしても)自律的にポドサイト障害を進める、いわゆるvicious cycleを形成して、これがFSGSの進展に新しい仮説をたてそれを証明したという、快心の仕事になりました。ASNでは高く評価され、これを“ポドサイトのドミノ倒し”と名づけ新しい概念として、少し苦労しましたが論文にしました。この研究を通して、FSGSの背景にTMAがあるだけではなく、TMAの背景にFSGSがあることもわかりました。(Koboyashi et al. Am J Physiol, Renal Physiol 2015)
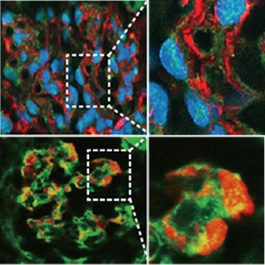
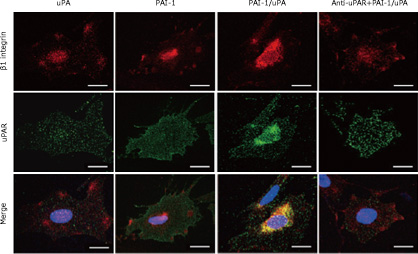
コントロールマウスでは、beta1インテグリンはポドサイトには認めないがNEP25マウスでは、ポドサイト内に取り込まれる。
(上:コントロール,下:NEP25,
赤:synaptopodin,緑:beta1インテグリン)
ポイントは左から3つめ。PAI-1/uPAの複合体で処理すると、細胞表面のインテグリンがuPARと同時にポドサイトの細胞内に取り込まれる。この現象は抗uPAR抗体でブロックされる。つまり、ポドサイトの基底膜接着を維持するbeta1インテグリンが細胞内に取り込まれた結果、ポドサイトは接着を失い剥離することを示している。
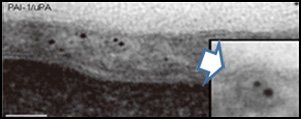
免疫電顕二重染色(小さな粒子beta1インテグリン、大きい粒子uPAR)これらが一緒にエンドサイトーシスの胞体内に取り込まれている決定的な写真(矢印)。
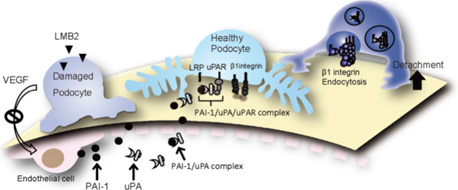
ポドサイトドミノ倒しのポンチ絵。
ポドサイトに障害が起きるとVEGF産生低下により、内皮細胞障害が起こり、内皮細胞はPAI-1を異常発現する。このPAI-1は血中でuPAと結合し、さらに隣の正常なポドサイトの表面にあるuPARとも結合することで、ポドサイトのインテグリンと複合体を形成する。これにより、インテグリンは細胞内にエンドサイトーシスされる。
④ FSGSで糸球体に泡沫細胞が出現する理由:ミステリーの解読
標本を観ていてどうしても知りたかったことがもう一つあります。FSGSでは何故糸球体に泡沫細胞がみられるのか?FSGSという病気の病理診断が曖昧にならざるを得ないところは、その疾患概念がどんどん変わっていることです。古典的な硬化の定義である血管の喪失と細胞外基質の増加にはあたらない新しい病変も認められています。
Collapsing FSGSなんてその典型でしょう。
これは、FSGS自体が臨床病理学的な概念なので、臨床的概念の拡大あるいは環境その他によるFSGSの多様性に伴い、FSGSと認知される病変の範囲がどんどん拡大しているためと考えられます。そういった多様なFSGS病変によく見られ、診断根拠とするものに、泡沫細胞があります。私の敬愛するD’Agati教授も、”it’s still mystery to me..“と言われています。私の恩師の渡辺照男先生は、動脈硬化の研究で有名で、動脈硬化が炎症によって起きるとのアイデアをいち早く提示されました。私は、動脈硬化にはほとんど興味のないままお世話になりましたが、それでも門前の小僧として泡沫細胞が動脈硬化病変に出てくる機序についてのいくつかの報告や基本的な知識は耳に入ってはいました。
IgG4関連腎症の仕事でご一緒し、指導を頂いた金沢大学の川野充弘先生から、一番優秀な若手を派遣していただき、この問題に取り組むことにしました。原玲史さんは、もともとベーシストのためなのか、仕事もリズミカルで大変早いのには驚きました。
泡沫細胞浸潤は、他の糸球体疾患でもみられますが、FSGSでの一番の特徴は、泡沫細胞の真上にあるポドサイトに障害が見えることです。つまり、FSGSがポドサイト病だとすれば、ポドサイトが障害を受けることが何らかの引き金になってマクロファージが局所で泡沫化する、という仮説をたてました。実際たいした高脂血症がなくても糸球体には泡沫細胞がやってきますから、高脂血症だけの問題でもなさそうです。また微小変化型ネフローゼ症候群(MCD)ではFSGSと同程度の高脂血症があってもこういう現象は起きないので、そのあたりを明確にしたいと考えました。これには、聞きかじった動脈硬化の知識が役に立ちました。
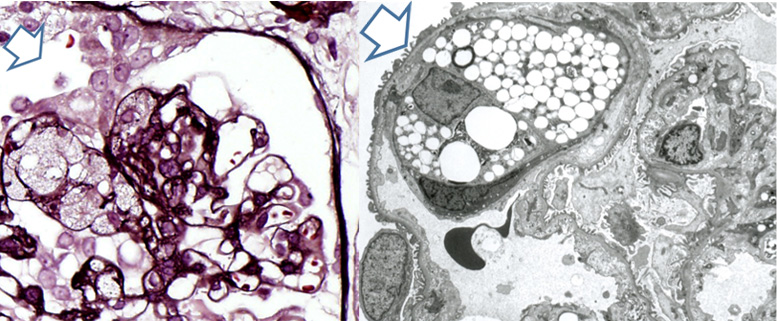
さて、この実験では複雑な手段を必要とすることは予測がついていましたが、原さんといろいろと議論するうちに、すべきことが次第に明確になってきました。実験はしばしば行き詰るものですが、予定調和のシナリヲを作ってから研究すると、その通りにならないときにどうしていいのかわからなくなります。いろいろと立ちはだかるハードルに、原さんがとても迅速に反応して、どちらに進むべきかを判断できる結果を常に用意してくれたおかげで楽しく研究が進みました。ここでもNEP25マウスのお世話になりましたが、LDL受容体欠損マウスと交配してダブルトランスジェニックマウスを作製し、またLDLKOマウスにアドリアマイシンFSGSも導入し検討しました。Cut the long story to shortは、英語でプレゼンをする日本人が最初に使いたがるフレーズですが、まあ要約すると、この研究の結論は、ポドサイト障害が起きるとおそらく局所の濾過障壁の機能低下、つまり透過性亢進(蛋白尿)あるいは内皮細胞障害により血中の脂質が糸球体の中に沈着しやすくなり、同時におそらく障害ポドサイト由来の活性酸素種(ROS)によりこの脂質が酸化され、それによって内皮細胞やメサンギウム細胞そしてマクロファージにサイトカインや接着分子などのネットワークが活性化するのではないかということです。この研究の面白く、また新しいところは、リピドミクスの手法を用いて、糸球体の中にたまっている脂質の種を解析し、2つの代表的過酸化脂質を同定したこと、そしてこの2つが、それぞれ異なった細胞に別々の作用をしていることを明らかにしたことです。考えてみれば、動脈硬化をプロトタイプとしてFSGSにおいていくつかの新しい発見がありました。現在投稿中なので、受理されたらデータを公表します。
⑤ 細胞間シグナル伝達は、細胞外基質を介する:形態学の苦手な領域
腎臓のような、多様な形質の細胞が複雑な構造をつくり、機能の恒常性を維持し、病的刺激に対して防御反応をする器官の場合、ひとつの細胞だけが周囲に構わず機能しているわけではありません。隣り合う細胞が社会を作り、お互いをよい状態にするべくシグナルを交換しています。このシグナル伝達は、どのように調節されているのでしょう?シグナル伝達の基本は、リガンドと受容体の結合によるスイッチ・オンです。
糸球体を観てみると、例えばポドサイトと内皮細胞、あるいはメサンギウム細胞の間には必ず細胞外基質がありますね。直接細胞同士がコンタクトを持っているわけではありません。メサンギウム細胞は、周囲に自分が産生した細胞外基質を持っており、内皮細胞とはほとんど直接の接点はありません。では、これらの細胞間の相互シグナルはどうやって伝わるのでしょうか?一般に受容体は細胞膜や核内に固定しており、リガンドは細胞外から受容体に近づいていくと理解されています。考えてみれば、リガンド自体には動くというモーターがないので、誰かに運んでもらわなくては隣の細胞の受容体には到達しないと考えられます。この運び手がサボってしまうと、いくら多くのシグナル分子が産生されても、隣の細胞にはシグナル伝達は起こらないということになります。このことは大変重要で、これまでの研究は、ひとつの細胞がシグナル分子を多く発現すれば、隣の細胞にその興奮がそのまま伝わる、とみなして論理が展開されていました。これは本当は正しくないことに気が付きました。
さて、前置きが長くなりましたが、糸球体を構築する細胞外基質には多くの種類がありますが、このシグナル伝達を調節していることがよく知られているのがヘパランサルフェートプロテオグリカン(HSPG)です。HSPGはメサンギウム基質にも、糸球体基底膜にも存在しますので、糸球体内の細胞間シグナル伝達を調節していると予測されます。これが本当に存在するのか、どのように機能しているのかという大きな問題に、院生の高島康利さんはあえてチャレンジしています。HSPGの中にあるサルファターゼという分子は、糸球体内で活躍するシグナルの代表である成長因子に結合する機能を持っています。つまりサルファターゼに結合した成長因子は、隣の細胞の受容体に到達することを一旦妨げられるということです。分子神経生物学の桝正幸先生との共同研究で、このサルファターゼを持っていないマウスでは、蛋白尿やメサンギウム増殖が起きることが初めて分かりました。その機序について現在研究を進めていますが、なかなか面白いことが分かってきています。
これも論文作成中なので、データは非公開です。
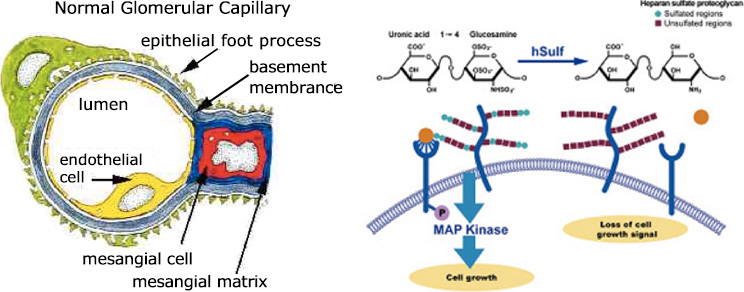
ポドサイト(緑)内皮細胞(黄色)、メサンギウム(赤)、細胞外基質(青)
3つの細胞は直接接点はありません。
必ず青い部分(細胞外基質)を介してクロストークをしていると考えられます。
右図はへパラン硫酸の図ですが、成長因子(オレンジの球)をサルファターゼがしっかりと保持し、受容体にバトンタッチしているのがわかります。
5.さらに新しい概念の提唱を目指して
結論が大体見えている研究をする時間も興味もなく、これからも新しい概念を提案する研究をしていきたいと思っています。現在取り組んでいるいくつかの研究については、まだ詳しくは説明できませんが、細胞間、あるいは臓器間のクロストークシステムを考える研究を始めました。これには、新しい技術が必要になってきますが、幸いなことに、大学内のいくつかの研究室と共同で基礎実験を始めました。腎臓は、全身の体液恒常性の維持を目的する臓器なので、左右の腎臓が相補的に協調して機能する必然があるのではないか、と考えています。これに関する課題とその検証方法を磨いていきながら、まだ誰も知らないことを少しずつでも明らかにしたいと思っています。
![]()