|
池田穰衛(東海大学・総合医学研究所・分子神経科学部門)
1. 研究の背景
私達は神経変性の素因とその分子機序を明らかにするために単一遺伝子の機能変異に基づく神経変性、即ち遺伝性神経変性疾患を対象として原因遺伝子の単離・同定ならびに当該遺伝子の機能解析を行っている。なかでも、遺伝子の機能喪失(loss
of function)を神経細胞死の背景とする劣性遺伝形式をとる神経変性疾患に着目して解析している。しかし、神経変性疾患に見られる細胞死は特定の神経に限られており、遺伝子機能レベルから神経細胞死の分子機序に迫るためには複数の神経変性疾患についてその原因遺伝子を同定し解析する必要がある。今回、私達が原因遺伝子を同定した家族性筋萎縮性側索硬化症2型(ALS2)も単一遺伝子のloss
of function を背景とする劣性遺伝形式をとる運動神経変性疾患の1つである。先にも、私達は劣性遺伝子疾患として知られている脊髄性筋萎縮症(SMA)の原因遺伝子を同定し機能解析を行っており、今回のALS2
遺伝子同定は神経細胞死の分子像に迫る新たな指標として期待している。
ALS2は一群の筋萎縮性側索硬化症(amyotrophic lateral sclerosis; ALS)の1つである。ALSは、大脳皮質錐体細胞から脊髄に至る上位運動ニューロンおよび脊髄前角細胞から筋に至る下位運動ニューロンが選択的に障害される進行性の神経変性疾患である。しかし、未だALSの素因は不明で、その有効な治療法・治療薬はない。ALS患者の大多数は孤発例で、家族性(遺伝性)ALSは全ALS発症例の5−10%である。遺伝性ALSについても複数例が報告されており、それらの症候や予後、遺伝子座も異なっていることからALSの発症には複数の因子が関わっていることが容易に想像できる。素因を明らかにするために、長年にわたって遺伝子性ALSを対象にその原因遺伝子の同定が試みられてきた。1993年にRosenら(Rosen,
D.R., et al.: Nature 362: 59-62, 1993)は常染色体優性遺伝形式を示す家族性ALS1型(ALS1)の原因遺伝子"SOD1"を同定することに成功した。SOD1遺伝子変異の発見は遺伝子レベルでの唯一の系としてALS分子病態研究を飛躍的に進展させた。しかし、大多数の孤発性ALS患者ではSOD1遺伝子変異が認められず、弧発性ALS発症の素因は別にあると考えられる。遺伝性ALSはもとより、ALSの大部分を占める孤発性ALSの発症素因を明らかにする手法としては、全てのALSに共通する"運動ニューロン障害・変性"の物質的背景が明らかである家族性ALS原因遺伝子機能の研究が最も有効な研究戦略である。私達が同定したALS2遺伝子は、ALS2遺伝子変異(loss
of function)が上位運動ニューロンの変性を誘起することから、ALS1原因遺伝子変異(SOD1遺伝子変異;gain
of function)に起因する下位運動ニューロンの変性と性質を異にしている。従って、ALS2遺伝子はALS発症分子機序の解明のための新たな指標分子と言える。
ALS2(若年発症型の家族性筋萎縮性側索硬化症2型)は臨床症候として3つのタイプに分別可能な常染色体劣性遺伝形式を示すチュニジアALS家系のうち"タイプ3"
がそれである(Ben Hamida, M., et al.: Brain 113: 347-363, 1990)。ALS2は上位運動ニューロンの変性に伴う四肢、顔面、咽頭筋の痙攣、構音障害等の偽球麻痺症状、および進行性の四肢筋萎縮等の諸症状を特徴とし、発症年齢が10歳未満と若年発症型で、病状の進行も極めて緩徐である。本疾患の初期症状は、若年発症型の原発性側索硬化症(juvenile
primary lateral sclerosis; JPLS)(Lerman-Sagie, T., et al.: J Child Neurol
11: 54-57, 1996)と極めて酷似しているが、進行性に四肢筋委縮を呈する患者が家系内に診られることから、下位運動ニューロン障害も併発していると推定される。1994年、Hentatiら(Nat
Genet 7: 425-428, 1994)は、上記のチュニジアALS2家系の遺伝子連鎖解析から、ALS2遺伝子座をヒト第2染色体長腕q33-q35のおよそ8センチモルガン(cM)の領域にマップした。次いで、Hoslerら(Neurogenetics
2: 34-42, 1998)はその候補領域を2つのDNA多型マーカーD2S116とD2S2237の間のおよそ1.7cM領域までに狭めた。私達は、S.
Schererらが調整した酵母人工染色体(YAC)、P1ファージ由来人工染色体(PAC)および大腸菌人工染色体(BAC)クローンを用いてALS2候補領域の詳細な物理地図(3Mb)と転写地図を作成した(Hadano,
S., et al.: Genomics 55: 106-112, 1999、71: 200-213, 2001)。引き続いて、私達はB.
Brownらから提供されたチュニジアとクウェートのALS2家系由来試料を用いてALS2遺伝子変異の同定に成功した。
2.研究成果
私達は、およそ3Mbの候補領域からALS2原因遺伝子を同定するために、cDNAクローニング、データーベース検索および塩基配列決定等の解析を行い、そこに存在する候補遺伝子を可能なかぎり多数同定した。その結果、ALS2候補領域には少なくとも42種類の転写配列(遺伝子)が存在することが明らかとなった。それら候補遺伝子の構造解析から、42個の遺伝子に由来する合計395個のエクソンを同定することに成功した。次に、各エクソンをPCR法により増幅した後、塩基配列を決定し、チュニジアALS2家系の患者と血縁関係にない正常人の配列を比較した結果、4種類の遺伝子(NOP5/NOP58、ALS2CR6、ALS2CR8、およびALS2CR9)のイントロン部分に合計6ヶ所、さらにエクソン部分に2ヶ所の疾患特異的塩基配列多型が見いだされた。これらのうち特に、新規遺伝子ALS2CR6の第3エクソン中に発見された1塩基対欠失変異(138delA;den
Dunnenらの変異配列命名法による表記、http://www.dmd.nl/mutnomen.html)は、ALS2CR6遺伝子のタンパク質コードフレームを壊すことから、有力なALS2遺伝子変異であると想定された。そこで、この遺伝子変異に関してさらに正常人533名に関して検索したが、いずれも変異は発見されなかった。一方、チュニジアの家系とは血縁関係にないクウェートのALS2/JPLS家系(Lerman-Sagie
T et al: J Child Neurol 11: 54-57, 1996)の試料を解析したところ、同一遺伝子の第5エクソンに2塩基対欠失変異(1425-1426delAG)が存在することが明らかとなった2)。さらに、Yangら(Yang,
Y., et al.: Nat Genet 29: 160-165, 2001)もサウジアラビアのJPLS家系において、同一遺伝子の第9エクソンに2塩基対欠失変異(1867-1868delCT)があることを報告した。これらの欠失変異は、いずれもALS2CR6遺伝子のタンパク質コードフレームを壊すことから(loss
of function)、これら遺伝子変異が劣性遺伝形質を説明し得るALS2遺伝子変異であるとの結論に至った。そして、ALS2CR6遺伝子を"ALS2遺伝子"と命名した。
ALS2遺伝子は、34個のエクソンからなる全長およそ80kbの遺伝子である。cDNAクローニングの結果、この遺伝子は6.5kbと2.7kbの2種類の転写配列を有すると推定された。そして全長ALS2転写配列は、推定1657アミノ酸からなる184
kDaのタンパク質(ALS2タンパク質)をコードしていると推定された。次に、ALS2遺伝子の正常組織における発現を解析するため、ノザンブロット解析を行った。その結果、およそ6.5kbと2.6kbの2種類のサイズのmRNAが検出され、cDNAクローニングの結果と矛盾の無い結果が得られた。遺伝子発現は、特に神経系、筋肉、および腎臓組織で強く、脳神経系では小脳における発現がもっとも顕著であった。さらに、ALS2患者末梢血細胞においても、同様に2種類のサイズの転写配列の発現が確認された。患者末血細胞由来のmRNAをRT-PCR法にて増幅し、その塩基配列を決定したところ、確かに患者からは欠失変異を含んだmRNAが発現していることが確認された。このことから、ALS2患者組織においても、変異mRNAを鋳型に異常欠失タンパク質が翻訳されている可能性が示唆された。
本研究ではさらに、ヒトALS2遺伝子のマウス相同遺伝子(Als2)を単離・同定した。Als2転写配列中には1,651アミノ酸をコードすると推定されるORFがあり、ヒトALS2遺伝子とマウスAls2遺伝子のORFにおける相似性は非常に高く、87%の塩基が同一であった。また、推定アミノ酸配列レベルでも94%の相同性を示し、ALS2遺伝子が哺乳動物においてよく保存されていることが明らかとなった。マウスAls2遺伝子の中枢神経系での発現をmRNA
in situ hybridization法により解析した結果、海馬錐体細胞、小脳プルキンエ細胞、大脳皮質、脊髄、扁桃体および顔面神経核等の神経細胞で強く発現していた。従って、ALS2/Als2遺伝子は、脳神経細胞全般にわたって発現していることが明らかとなった。従って、ALS2遺伝子変異により引き起こされる選択的神経細胞死に関しては、ALS2遺伝子に加え、さらに別の因子の関与が想定される。
次に、ALS2遺伝子にコードされているALS2タンパク質の機能を推定するため、推定アミノ酸配列のホモロジー検索およびドメイン・モチーフ検索を行った。ALS2タンパク質には、グアニンヌクレオチド交換因子(guanine
nucleotide exchange factor; GEF)の特徴的ドメイン構造が複数存在することが明らかになった(regulator
of chromosome condensation (RCC1)-like domain; RLD、Dbl homology/pleckstrin
homology; DH/PH、およびvacuolar protein sorting 9; VPS9)(図)。RCC1はRan GTPase
の活性化を触媒するGEFであり、微小管アセンブリ調節によりクロマチン凝集に関与する分子である(Carazo-Salas, R.E., et
al.: Nature 400: 178-181, 1999)。また、RCC1様配列は色素性網膜炎の原因遺伝子RPGRおよび線虫のRPM-1遺伝子産物にもみられ、それぞれ網膜細胞および神経細胞シナプス終末において神経細胞の維持あるいは可塑性に関連した働きをしていると想定されている。さらに、RCC1様配列を有するヒトp532タンパク質は、RanよりむしろARF1、Rab3およびRab5に対するGEF活性を有すると報告されている(Rosa,
J.L., et al.: EMBO J. 15: 4262-4273, 1996)。一方、DH/PHのタンデム構造は、Rho タイプのGEFに典型的なドメイン構造であり、様々なシグナル伝達系、神経細胞形態形成、細胞膜・細胞小器官輸送、アクチン細胞骨格構造の調節機能があると想定されている(Luo,
L.: Nat Rev Neurosci 1: 173-180, 2000)。さらに、VPS9はRab5 GTPaseを活性化するGEFに共通したドメインであり、エンドサイトーシス、特にエンドゾームにおける小胞融合に関与していることが明らかにされている(Zerial,
M. & McBride, H.: Nat Rev Mol Cell Biol 2: 107-117, 2001)。また、DH/PHの下流に存在する7つのMORNモチーフは、細胞膜への結合に関与するアミノ酸配列である(Takeshima,
H., et al.: Mol Cell 6: 11-22, 2000)。以上の知見から、ALS2タンパク質は、低分子量GTPase活性調節に関連したシグナル伝達系において役割を担っている可能性が高く、神経細胞ではそれらの機能を通して、微小管アセンブリ、膜構造調節あるいは軸索輸送などの神経細胞機能構造構築に関与していると推定される。しかし、これらの分子機能はいずれもアミノ酸配列におけるホモロジーから予測されたものであり、実際のタンパク質機能の解明に関しては今後の研究課題である。
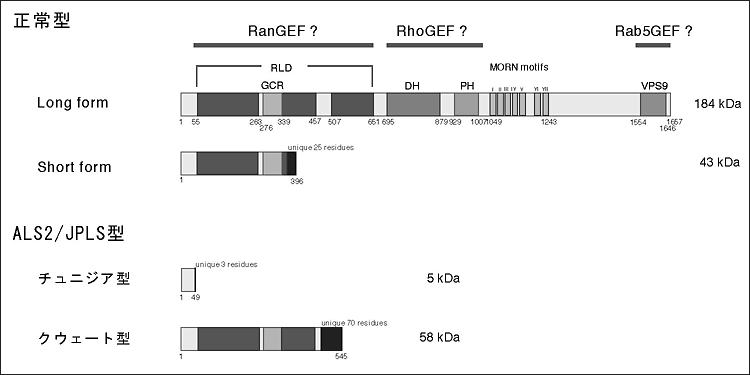 |
図 「ALS2タンパク質におけるドメイン・モチーフ構造および推定変異タンパク質の構造」
RLD; Rcc1-like domain, GCR; glucocorticoid receptor, DH; Dbl homology
domain, PH; pleckstrin homology domain, MORN; membrane occupation
and recognition nexus, VPS9; vacuolar protein sorting 9 domain. |
3.おわりに
ALS2は、ALS2遺伝子の"機能損失変異"によりALS2タンパク質の本来発揮すべき機能が損なわれ、それにより運動ニューロン機能障害および細胞死が引き起こされていると考えられる。換言すれば、ALS2タンパク質は神経細胞の生存・維持にかかわる必須因子である。従って、この新規ALS関連タンパク質の正常な分子機能を知ることは、ALS2の分子発症機序の解明にとどまらず、ALSの分子病態解析、運動ニューロンの生存・維持機構、および細胞機能障害・細胞死の分子カスケードを解明する上で不可欠であると考えられる。現在、私達はALS2遺伝子産物の生化学的・細胞生物学的手法による分子機能解析、およびALS2モデルマウス(Als2ノックアウトマウス)の作製を開始している。このような実験を通して今後ALS2タンパク質の機能が明らかにされることが期待される。そして、新たな家族性ALS遺伝子の同定と機能解析の研究の積み重ねが、近い将来、ALSの分子病態の解明、さらには有効な治療法・治療薬の開発に結びつくことを期待している。
本稿に紹介したALS2研究は科学技術振興事業団日加国際共同研究「神経遺伝子」プロジェクト(1996年1月〜2000年12月)の一環として行ったものである。
劣性遺伝病の原因遺伝子ハンティングは物理的煩雑さは然ることながら、運・不運共々一筋縄では行かない領分のものである。ALS2遺伝子のハンティングに際して、必然的な無為・無駄を躊躇していたが、本論文の筆頭著者秦野伸二君の「それでもヤッテみましょうよ」の一言が無ければ今回の成功はありえなかったでしょう。
|
