| 郭 伸 (東京大学・大学院医学系研究科・脳神経医学専攻・神経内科学教室)
1.研究の背景
筋萎縮性側索硬化症 (ALS)は、1872年〜1874年にわたるJean-M Charcotによる連続系統講義の中で疾患概念として提唱された、骨格筋を支配する運動ニューロンが選択的かつ系統的に変性脱落することにより、呼吸筋を含むほぼ全身の筋萎縮を来す進行性の神経難病である。ALSの有病率は世界的に10万人あたり2〜8人で、本邦の患者数は5〜6千人になると推定される。身体的な障害が重い上に、末期まで意識、知能が保たれることもあり、精神的な負荷も大きく、また、介護に当たる家族も長期にわたり他に類を見ない物心面での負担を強いられることから、病因の解明、特異的治療法の開発は、社会的、人道的にも急務である。
疾患概念の確立以来130年以上が経過し、様々な疾患研究アプローチにもかかわらず、病因はおろか、運動ニューロンの選択的細胞死のメカニズムに関してすら未解明であった。5-10%を占める家族性ALSのうち、3疾患(ALS1,
ALS2, ALS4)では遺伝子異常(SOD1, ALS2, senataxin)が同定されたものの、なぜ運動ニューロンが死ぬのかというメカニズムは依然として解明されていない。大多数を占める孤発性ALSについては、様々な病因仮説のなかで現在最も有力なものがグルタミン酸受容体であるAMPA受容体を介した興奮性神経細胞死仮説である。錐体路はグルタミン酸を神経伝達物質として用いており、脊髄運動ニューロンはこの興奮性入力を豊富に受けている。AMPA受容体は、脊髄運動ニューロンの細胞死に関与し、Ca2+透過性変化が注目されてきた。
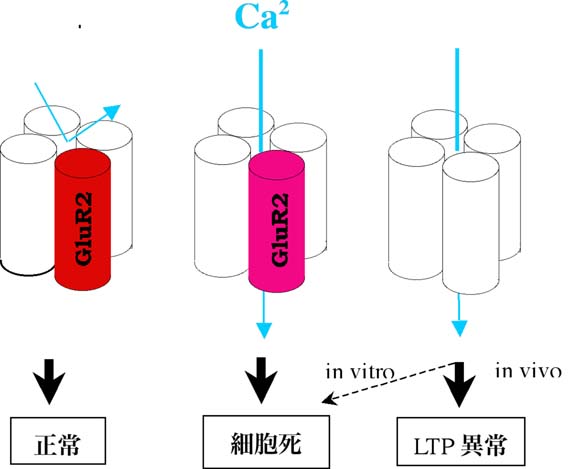
AMPA受容体は、GluR1-GluR4サブユニットで構成されるheteromericな四量体で、このうちチャネルのCa2+透過性を決定するのはGluR2である。GluR2を含むAMPA受容体はCa2+透過性は低く、1つも含まないと透過性が高い(図1)。GluR2のこのような性質は、第2膜ドメインにあるQ/R部位を、他のサブユニットはグルタミン(Q)が占めているのにアルギニン(R)に置き換わっているためである。このアミノ酸置換は転写後にRNA編集を受けることにより遺伝子にコードされているQがRに変わるもので、哺乳類の神経細胞ではほぼ100%
Rを発現している。未編集のGluR2Qを含むAMPA受容体はCa2+透過性が高いので、RNA編集率の低下は、編集型GluR2Rの減少と同様、AMPA受容体のCa2+透過性を高める。
|
図1.AMPA受容体とGluR2サブユニット
AMPA受容体はサブユニットGluR1〜GluR4による4量体。編集型GluR2(赤)がサブユニットに入っているとCa2+非透過性だが(左)、入っていないAMPA受容体は透過性である(右)。未編集型GluR2(桃色)が入っていてもやはりCa2+透過性になる(中)。
|
我々は、ALS脊髄運動ニューロンのGluR2 mRNA発現量に有意な減少を認めないことを単一神経細胞レベルで確認した上で4)、果たして死に行く運動ニューロンで未編集GluR2
mRNAが増えているかどうかを確認する実験を行った。
非NMDA型グルタミン酸受容体を介した細胞死が脊髄ニューロンの細胞死と関係があることを1980年代末から見いだしており1),5-10)、動物実験を積み重ねるうちに、ALS脊髄運動ニューロンにおけるAMPA受容体サブユニット分子異常を突き止めることができた。今回の成果が研究の継続により初めてもたらされたものであると自負しており、研究方向転換の勧めに逆らって意地を通した経緯を考えると感慨深いものがある。
2.研究の概要
我々が得ていたRNA編集低下は脊髄前角組織でのものであったため12)、個々の運動ニューロンでの異常を反映したものであるかどうかは結論できず、運動ニューロン組織での検討が必須であった。
単一ニューロンでの検討を可能にしたのは、恩師である金澤一郎名誉教授が開発したエキシマレーザー光を用いたミクロディセクターによっている。現在でこそ数種類が商品化されているが、1997-8年当時は手探りの状態であり、このシステムの立ち上げには、現在日赤医療センター神経内科で活躍している橋田秀司博士の並ならぬ努力に負うところが大きい。本研究では、単一運動ニューロン組織をヒト剖検脳・脊髄よりミクロディセクターを用いて切り出し、その微少組織からRT-PCRによりGluR2
mRNA由来のcDNAを増幅して、編集部位を認識する制限酵素Bbv-1処理により生ずる消化断片の定量により編集率を算出した。ニューロンはGluR2
mRNAを比較的豊富に発現していることが分かり、50%以上80%近くのニューロンでGluR2 mRNAを検出することができた。
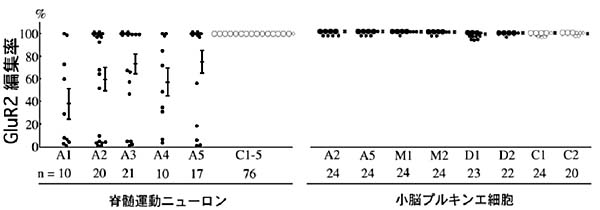
その結果は、図2に示すように、正常対照群5例の運動ニューロン76細胞では、全例GluR2 Q/R部位はRNA編集されていたのに対し、ALS群5例78細胞では0-100%とばらつき、平均値は38-75%と低下していた。部位選択性を明らかにする目的で、ALSの小脳プルキンエ細胞、疾患特異性を明らかにする目的で、小脳変性を来す多系統萎縮症、dentato-rubural
pallidoluysian atrophy (DRPLA)の同細胞における編集率を測定し、正常対照同様ほぼ100%に保たれていることを確認した。したがって、ALS脊髄運動ニューロンで認められるRNA編集異常は、疾患特異的・細胞選択的な分子変化であるといえる。ニューロン組織以外のコンタミはMAP2、GFAPに対するPCRでチェックし、ニューロピル組織からはGluR2
mRNAが検出できないことも確かめた。
|
図2.単一神経細胞におけるGluR2
Q/R部位RNA編集率
各点 (大きな点は5細胞、小さな点は1細胞)は、ALS (A1-A5)、コントロール (C1-C5)の単一脊髄運動ニューロン、およびALS、多系統萎縮症
(M1, M2)、DRPLA (D1, D2)、コントロールの単一小脳プルキンエ細胞におけるGluR2 Q/R部位のRNA編集率を表している。平均値
± 標準誤差、解析した細胞数 (n):C1; 28, C2; 12, C3; 13, C4; 12, C5; 11。
|
3.研究成果の意義
1)細胞死とRNA編集異常:運動ニューロン選択性、疾患特異性
ニューロンにおけるGluR2のRNA編集は、発生初期から99%以上に保たれている2)。GluR2のQ/R部位をゲノムレベルでR(すなわち編集型)を発現するトランスジェニックマウスの表現型は正常であるが、同部位にQを発現する変異マウスは、ホモ接合体、ヘテロ接合体とも、生後20日以内に死亡した。この変異マウスの海馬錐体細胞ではAMPA受容体のCa2+透過性が亢進しており、Q/R部位のRNA編集が神経細胞にとって非常に重要な修飾であり、未編集型GluR2は発生段階にかかわらず障害性に働くので、編集型GluR2のみを発現するようにプログラムされていると考えられる。逆に、このプログラムがうまく働かないと、細胞死、個体死を引き起こす。
ALS疾患特異性に関しては、今回発表したデータの他に、ハンチントン病の線条体やアルツハイマー病の大脳皮質などの組織レベルで、ALS以外の神経変性疾患では病理変化の強い部分でも同部位のRNA編集率は低下していないことを示した11)。また、悪性グリオーマの低下が報告されているが、正常対照ヒト白質でも同程度の低下がみられ3)、グリア細胞ではAMPA受容体のCa2+透過性が細胞死を引き起こさないこともあり、病因的意義はALSにおけるものとは異なるといえる。
部位選択性に関しては、GluR2 Q/R部位をアスパラギン(N)に置換したminigene GluR2(N)を挿入したマウスで、生後12ヶ月で遅発性に脊髄運動ニューロンが変性し、AMPA受容体のCa2+透過性も亢進していたことが報告されている。このマウスでは、GluR2(N)はGluR2
全体の20数%の発現に過ぎないが、比較的少量の未編集型GluR2がAMPA受容体の機能に変化を及ぼす因子として、GluRのQ/R部位が、AMPA受容体アセンブリー、
輸送メカニズムを制御し、Q型が機能的AMPA受容体を形成しやすいことが示されている。さらに、運動ニューロンのGluR2 mRNA発現が低いこと4)を考えると、RNA編集率の低下がCa2+透過性AMPA受容体形成に及ぼす影響は運動ニューロンでより大きいといえる。
このように、RNA編集という生物現象が破綻することにより、活性分子であるAMPA受容体に細胞死を引き起こす機能異常が生じることが生物学的に明らかにされており、この異常が特定の疾患に特異的に生じている例は稀少である。機能異常が明らかであれば、治療法開発への道はより近いといえる。
2)RNA編集13)
一般に、遺伝子からmRNAが形成される過程で、遺伝情報が書き換えられることをRNA editing (編集)と呼ぶ。RNA編集は、転写後のコドンの挿入削除と、転写後のコドン置換がある。後者には、シトシン(C)→ウラシル(U)と、アデノシン(A)→イノシン(I)の二種類が知られ、特にA→I置換は中枢神経系で最も活発に生じていることが、種を超えて確認されている。
A→I RNA編集には、多塩基置換型(promiscuous editing)と一塩基置換型があり、特に後者は、翻訳されるアミノ酸配列が変わり、生理学的性質も変化するという点から極めて重要である。既知の編集部位は、今のところ神経伝達物質受容体やイオンチャネルに集中しているが、多数の未知の編集部位が存在していると考えられている。
A→I RNA編集は、adenosine deaminases acting on RNA(ADARs)と呼ばれる酵素によって触媒される。哺乳類ではADAR1〜ADAR3の3種類が知られており、これらは2本鎖RNA結合部位と編集触媒部位を共通の構造として持つ。GluR2
mRNA Q/R 部位はADAR2により触媒されることが動物脳で示され、ヒト脳でも同様であると考えられる3)。
ADARsが、1本鎖RNAであるmRNAを編集できるのは、編集部位を含んだエクソンに隣接したイントロンに、exon complementary
sequence(ECS)と呼ばれる不完全相同配列が存在しており、pre-mRNAの段階でECSと部分的な2本鎖が形成されることによる。置換されたIは、翻訳段階でグアノシン(G)と同等であると見なされる。GluR2
mRNAのみがECSをもつため、RNA編集を受ける。
ADAR2のノックアウトマウスはGluR2 Q/R部位の編集率が10%以下に著減し、GluR2のRNA編集を欠損させた変異マウスと同じく幼弱のうちに死亡してしまうが、野生型GluR2をコードする遺伝子の導入により、細胞死も個体の幼弱死も回避できる。このことは、GluR2
mRNA編集の低下自体が細胞、個体の生存に関わることを示しており、ALSの脊髄運動ニューロンに特異的にみられたGluR2 Q/R部位RNA編集異常が直接的なニューロン死の原因であることを意味する点でALSの病因との強い関連を示唆する。
4.課題・展望
以上のことから、ALSにおけるGluR2 Q/R 部位のRNA編集異常は、編集酵素ADAR2の活性低下によると推定される。そのメカニズムとして、ADAR2発現量がひとつの因子であることをつかんでいる3)ほかにも、活性を低下させる分子修飾、促進因子の欠如、阻害因子などが想定される。特異治療の開発に向け標的が絞られたことで、一刻も早く治療薬を開発し患者さんへ還元したいと考えている
今回の発表ではいろいろな勉強をさせてもらった。成果のなかなか出ない研究を続けることに対して、発表前には、以前やっていた研究を続けていたら良かったのにという好意からの意見、(内容ではなく)やり続けることの意義を認めた励まし、臨床医がやるいい加減な実験のためのPCRエラーだろうという無視、などの反応であったが、今回、「先生が長年言っていたことは本当だったのだね」と180度評価が変わったことにはインパクトの大きさを実感したものである。考えてみるとこの研究紹介も内容で選ばれたのではなく、掲載された雑誌が選択基準のようであり、研究評価を外部委託していることになる。患者さんからの反響は大きく、新規治療法はまだかとの多数の問い合わせのほか、研究費を寄付してくださる方もあった。
研究を継続できた背景には、研究材料にとご遺体を提供してくださった患者さんのご遺志、鳴かず飛ばずの臨床医の研究にそれなりの研究費を付け続けてくださった慧眼の諸先輩に負うところが大きくこの場を借りて感謝したい。
文献
1) Hirata, A., et al. Brain Res. 771, 37-44.
(1997).
2) Kawahara, Y., et al. Brain Res Dev Brain Res
148, 151-5 (2004).
3) Kawahara, Y., et al. Eur J Neurosci 18,
23-33 (2003).
4) Kawahara, Y., et al. J Neurochem 85, 680-689
(2003).
5) Kwak, S., et al. Exp. Neurology 116, 145-155
(1992).
6) Kwak, S., et al. Neuroscience 73, 687-695
(1996).
7) Kwak, S., et al. J. Neurol. Sci. 129 (Suppl),
99-103 (1995).
8) Kwak, S., et al. Brain Res. 702, 61-71
(1995).
9) Nakamura, R., et al. Brain Res 654, 279-285
(1994).
10) Shinozaki, H., et al. Brain Res 503, 330-333
(1989).
11) Suzuki, T., et al. Nippon Yakurigaku Zasshi
122, 515-26 (2003).
12) Takuma, H., et al. Ann Neurol 46, 806-815
(1999).
13) 河原行郎、郭 伸 Clinical Neuroscience 22, 250-251
(2004).
|